Institution: Weill Cornell Medical College
Additional authors:Attilio Orazi
Session: AML secondary to myeloproliferative neoplasms and other types of disease progression in MPN
HISTORY
The patient is a 72 year old man who was reportedly diagnosed with essential thrombocythemia in 2001 (slides were not available for review). At the time of diagnosis his platelet count was ~1.2 million and the clinical picture was complicated by a transient ischemic attack. The patient was treated with anagrelide in combination with hydrea until October 2009 when he developed anemia and became transfusion-dependent. A bone marrow performed in December 2009 showed evidence of a chronic myeloproliferative neoplasm (MPN) with 1 to 2+ reticulin fibrosis and no increase in blasts. A bone marrow examination performed in February 2010 showed increased fibrosis and 10% blasts by flow cytometry. The patient was treated with 4 cycles of decitabine, and a follow-up bone marrow examination in May 2010 reported a markedly hypocellular bone marrow with 1+ fibrosis and without increased blasts on biopsy or aspirate smear. Subsequently, the patient did well until he again became transfusion-dependent in November 2010. A bone marrow biopsy showed accelerated phase MPN with 16% blasts and 3+ reticulin fibrosis. The patient was treated with an investigational drug TG02 (oral multi-kinase inhibitor), however he remained transfusion-dependent and had detectable circulating blasts in peripheral blood (1-9% of all cells). A bone marrow biopsy in July 2011 showed 15% blasts, 3+ fibrosis and a new cytogenetic abnormality. In September 2011, the patient was admitted due to acute renal insufficiency and altered mental status. A bone marrow biopsy showed 24% blasts. He was treated with 7+3 induction regimen, complicated by tumor lysis syndrome, worsening renal failure, respiratory failure and sepsis. Chemotherapy was discontinued. The patient expired in November 2011.
DETAILS
All bone marrow biopsies were from the posterior iliac crest and were fixed in Bouin's solution. The cellularity was variable, but overall was normal for patient's age (~30%). The biopsies showed increased megakaryopoiesis characterized by the presence of highly atypical pleomorphic forms ranging in size from small nonlobated or hypolobulated to large hyperlobulated megakaryocytes with hyperchromatic nuclei. In areas, the megakaryocytes displayed a diffuse sheet-like growth pattern. A variably increased number of blasts was seen. Maturing myeloid and erythroid precursors were markedly decreased in number. A special stain for reticulin showed marked (3+) reticulin fibrosis.
IMMUNOHISTOCHEMISTRY AND FLOW CYTOMETRY
Flow cytometric analysis identified a blast population that expressed CD34 (dim), CD117, CD13, CD33, HLA-DR, CD36, CD56 and CD41. Immunohistochemical staining revealed an increased number of CD34(-), CD117(+) myeloid blasts. MPO highlighted rare left-shifted myeloid cells, Glycophorin C was positive in rare scattered erythroid cells. CD42b and CD61 highlighted a marked increase in the number of megakaryocytes, micromegakaryocytes and megakaryoblasts. These cells focally form large aggregates. TdT, CD56 and CD99 were negative.
CYTOGENETIC FINDINGS
46,XY until July 2011, when the patient acquired a new translocation: 46,XY,der(6)t(1;6)(q21;p21)
MOLECULAR FINDINGS
Negative for JAK2, FLT3, NPM1 and CEBPA mutations
INTERESTING FEATURES
Disease progression is uncommon in patients with essential thrombocythemia. Since we did not have the opportunity to review the patient’s diagnostic bone marrow biopsy, there is a possibility that based on the WHO criteria the correct initial diagnosis was that of early primary myelofibrosis with thrombocytosis. Translocation t(1;6)(q21;p21) appears to be highly specific to patients with myelofibrosis (Dingli D et al, Br J Haematol 2005). In this case, it was acquired in the course of disease progression from accelerated phase to the blast phase. While de novo acute megakaryoblastic leukemia is very rare, megakaryoblastic transformation of myeloproliferative neoplasms likely occurs more frequently (Hernandez JM et al, J Clin Pathol 1992; Georgii A et al, Leuk Lymphoma 1996; Vianelli N et al, Haematologica 1996; Orazi A et al, Mod Pathol 2005).
PROPOSED DIAGNOSIS
Myelofibrotic myeloproliferative neoplasm, megakaryoblastic blast phase.
CONSENSUS DIAGNOSIS
Myeloproliferative neoplasm with myelofibrosis [post ET versus PMF], accelerated phase progressing to megakaryoblastic blast phase, with der(6)t(1;6)(q21;p21)
| The bone marrow is hypocellular for age with evidence of severe fibrosis and osteosclerosis |  |
| Other areas are more cellular and contain an increased number of atypical megakaryocytes | 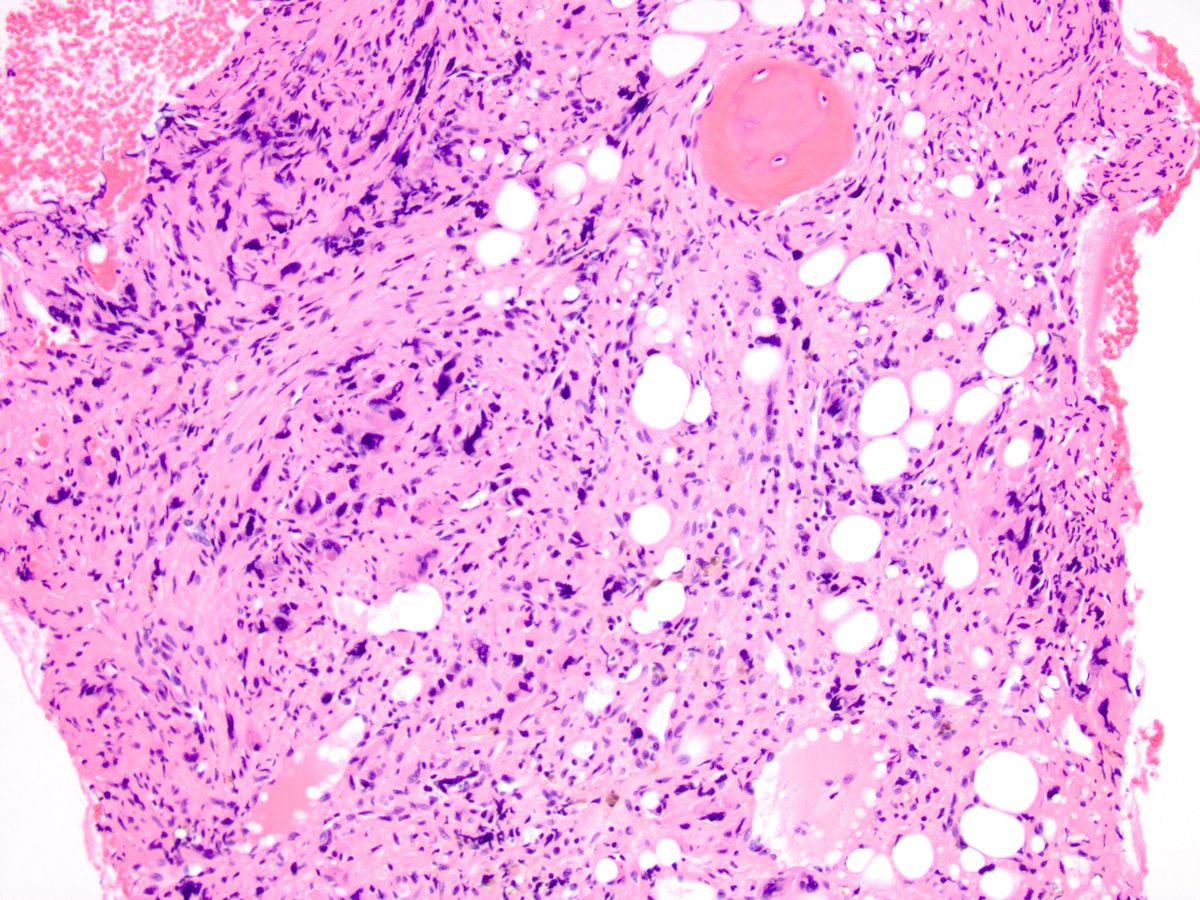 |
| A high-power image shows a marked megakaryocytic proliferation with abnormal-appearing megakaryocytes, micromegakaryocytes and megakaryoblasts. Normal maturing hematopoiesis is virtually absent. |  |
| Immunohistochemistry for CD42b highlights sheets of abnormal megakaryocytes |  |
| Immunohistochemistry for CD61 shows increased number of megakaryocytes and megakaryoblasts |  |
| Circulating megakaryoblasts are noted in peripheral blood |  |