Institution: UT Southwestern Medical Center at Dallas, Dallas, TX
Additional authors:Jacqueline Emmons MD, Prasad Koduru PhD
Session: AML with myelodysplasia-related changes
HISTORY
The patient is a 60 year old woman who presented to the hematology/oncology clinic with a chief complaint of chronic fatigue and dyspnea on exertion that developed over the previous ten months. Her past medical history was notable for chronic kidney disease and lung cancer treated by resection only. There was no history of cytotoxic chemotherapy or radiation. Macrocytic anemia and thrombocytosis were noted in the patient's chart on routine complete blood counts over a course of one year prior to presentation. Automated platelet counts ranged from 449-821 x 10(9)/L (reference range 174-404 x 10(9)/L). Hemoglobin averaged 9.5 g/dL (reference range 12.1-16.1 g/dL) with mean corpuscular volumes of 97.5-108.9 femtoliters (reference range 76.2-98.6 femtoliters). White blood cell count was normal with an unremarkable differential count. Vitamin B-12, folate, and iron studies excluded nutritional deficiency. The patient's medications were not believed to be contributing to her symptoms or abnormal cell counts. A peripheral blood specimen was sent for molecular testing that was reported positive for JAK2 V617F mutation measuring 0.1% of total JAK2 DNA. A bone marrow biopsy was performed to ascertain the source of anemia and thrombocytosis.
DETAILS
A biopsy of the bone marrow was taken from the left posterior superior iliac spine. Bone marrow aspirate smears were made and a trephine core biopsy was fixed in B-5 for permanent paraffin sections. A portion of the aspirated marrow was sent for flow cytometry, molecular testing for JAK2 V617F mutation, conventional cytogenetic studies, and fluorescence in situ hybridization (FISH) with probes specific for myelodysplastic syndromes (MDS).
An accompanying peripheral blood smear confirmed a macrocytic anemia with anisocytosis, occasional macrocytes, and rare teardrop cells. Platelets were increased, appropriately granular, and included scattered giant forms. White blood cells were normal in number and exhibited unremarkable morphology; no blasts or dysplastic features were appreciated.The bone marrow aspirate smear was cellular with trilineal hematopoiesis. Megakaryocytes appeared increased and included frequent small and/or monolobated forms. There was a normal complement of erythroid precursors with progressive maturation and rare nuclear budding; otherwise there was no distinct dyserythropoiesis. Granulocyte precursors exhibited progressive maturation and unremarkable morphology. Blasts were not increased in the manual differential cell count.The core biopsy and clot sections indicated a bone marrow cellularity of approximately 60%. Megakaryocytes were increased, present in clusters, and included frequent monolobated forms. Granulocyte and erythroid precursors were present in normal numbers and exhibited progressive maturation. Iron stain of the aspirate smear confirmed the presence of storage and sideroblastic iron. Ring sideroblasts were not identified.IMMUNOHISTOCHEMISTRY AND FLOW CYTOMETRY
Immunohistochemical stains were not performed. Flow cytometry of the bone marrow aspirate provided no definitive immunophenotypic evidence of a hematolymphoid malignancy. Myeloblasts were not increased or immunophenotypically aberrant. Granulocytes and monocytes did exhibit partial expression of CD56.
CYTOGENETIC FINDINGS
Conventional cytogenetics identified an abnormal karyotype containing a deletion in the long (q) arm of chromosome 5 in fifteen of 20 cells examined. A normal female chromosome complement was observed in 5 cells.
FISH with probes specific for MDS showed evidence of a deletion in the long arm of chromosome 5 in 61.5% of 200 cells examined.MOLECULAR FINDINGS
Molecular testing of the bone marrow aspirate once again identified a JAK2 V617F mutation measuring 0.8% of total JAK2 DNA.
INTERESTING FEATURES
The clinical history of macrocytic anemia and thrombocytosis, the presence of hyperplastic dysmegakaryopoiesis with minimal erythroid dysplasia, and the accompanying cytogenetic findings are all indicative of the myelodysplastic syndrome with isolated deletion of 5q. JAK2 V617F mutations are uncommonly found in conjunction with isolated deletion 5q, but this phenomenon has been reported. It is unknown what, if any, effect a JAK2 mutation has on the pathogenesis or clinical course of myelodysplastic syndrome associated with isolated deletion 5q.
PROPOSED DIAGNOSIS
Myelodysplastic syndrome with isolated deletion 5q. A concomitant JAK2 mutation is noted, but the significance of this finding is not certain.
CONSENSUS DIAGNOSIS
Myelodysplastic syndrome with isolated del(5q)
| Peripheral blood 20x showing a macrocytic anemia and thrombocytosis |  |
| Bone marrow aspirate 10x showing an increase in megakaryocytes with several monolobated forms |  |
| Bone marrow aspirate 20x showing monolobated megakaryocytes and progressive maturation in granulocyte and erythroid precursors without distinct dyspoiesis |  |
| Bone marrow core biopsy 10x showing increased megakaryocytes with many monolobated forms |  |
| Bone marrow core biopsy 20x showing many monolobated megakaryocytes and progressive maturation in granulocyte and erythroid precursors |  |
| Conventional karyotype identified deletion in the long (q) arm of chromosome 5 | 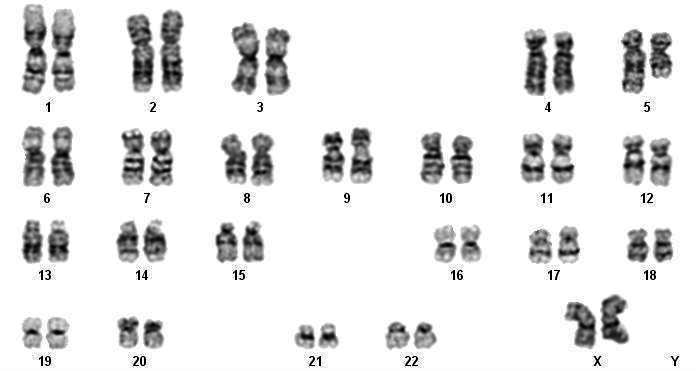 |
| FISH for EGR1 on chromosome 5: Normal signal on left, abnormal signal on right indicating 5q deletion | 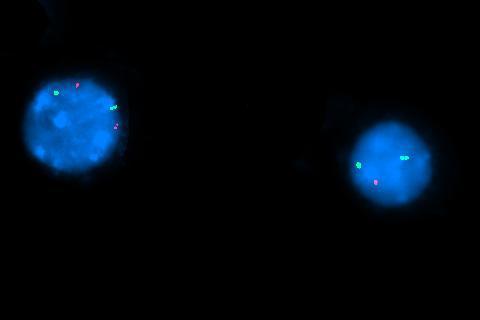 |